Ellappan Babu, Ph. D, Sabarish Ramachandran, Ph. D, Veena CoothanKandaswamy, Ph. D., Selvakumar Elangovan, Ph. D, Puttur D. Prasad, Ph. D, Vadivel Ganapathy, Ph. D, and Muthusamy Thangaraju, Ph. D.
Department of Biochemistry and Molecular Biology, Medical College of Georgia, Georgia Health Sciences University, Augusta, Georgia, USA
Users may view, print, copy, download and text and data- mine the content in such documents, for the purposes of academic research, subject always to the full Conditions of use:
http://www.nature.com/authors/editorial_policies/license.html#terms
Correspondence: M. Thangaraju, Ph. D., Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta, GA 30912, USA., [email protected].
Conflict of interest: The authors declare that there is no competing financial interest in relation to the work described in this manuscript.
Published in final edited form as: Oncogene. 2011 September 22; 30(38): 4026–4037. doi:10.1038/onc.2011.113.
Keywords: SLC5A8; dichloroacetate; anticancer drug; Warburg effect; pyruvate dehydrogenase kinase; mitochondrial oxidation in cancer
Abstract
There has been growing interest among the public and scientists in dichloroacetate as a potential anticancer drug. Credible evidence exists for the antitumor activity of this compound, but high concentrations are needed for significant therapeutic effect. Unfortunately, these high concentrations produce detrimental side effects involving nervous system, thereby precluding its use for cancer treatment. The mechanistic basis of the compound’s antitumor activity is its ability to activate pyruvate dehydrogenase complex through inhibition of pyruvate dehydrogenase kinase. Since the compound inhibits the kinase at micromolar concentrations, it is not known why therapeutically prohibitive high doses are needed for suppression of tumor growth. We hypothesized that lack of effective mechanisms for the entry of dichloroacetate into tumor cells may underlie this phenomenon. Here we show that SLC5A8 transports dichloroacetate very effectively with high affinity. This transporter is expressed in normal cells, but the expression is silenced in tumor cells via epigenetic mechanisms. The lack of the transporter makes tumor cells resistant to the antitumor activity of dichloroacetate. However, if the transporter is expressed in tumor cells ectopically, the cells become sensitive to the drug at low concentrations. This is evident in breast cancer cells, colon cancer cells, and prostate cancer cells. Normal cells, which constitutively express the transporter, are however not affected by the compound, indicating the tumor cell-selective therapeutic activity. The mechanism of the antitumor activity of the compound is still its ability to inhibit pyruvate dehydrogenase kinase and force mitochondrial oxidation of pyruvate. Since the silencing of SLC5A8 in tumors involves DNA methylation and its expression can be induced by treatment with DNA methylation inhibitors, our findings suggest that combining dichloroacetate with a DNA methylation inhibitor would offer a means to reduce the doses of dichloroacetate to avoid detrimental effects associated with high doses but without compromising antitumor activity.
INTRODUCTION
Dichloroacetate is currently used for the treatment of congenital lactic acidosis (Stacpoole et al., 2003, 2008). The therapeutic efficacy of this drug is due to its ability to activate pyruvate dehydrogenase complex (PDC) in mitochondrial matrix. However, the enzyme complex is not the direct target for the drug. Dichloroacetate is an inhibitor of pyruvate dehydrogenase kinase (PDK), which phosphorylates the E1α subunit of PDC and inactivates the complex (Stacpoole et al. 2003, 2008). By inhibiting PDK, dichloroacetate prevents the phosphorylation of E1α and thus maintains PDC in active form. The drug-induced activation of PDC facilitates mitochondrial metabolism of pyruvate. Since cytosolic pyruvate can either be transported into mitochondria and metabolized or be converted into lactate in cytoplasm by lactate dehydrogenase, activation of PDC by dichloroacetate and the resultant metabolism of pyruvate within mitochondria shifts the equilibrium between pyruvate and lactate towards pyruvate. This promotes conversion of lactate into pyruvate, thus reducing lactate levels.
There has been considerable interest in recent years in dichloroacetate as a potential anticancer drug (Michelakis et al., 2008). The notion that this drug may have ability to kill tumor cells has a rational basis. Tumor cells derive most of their energy from aerobic glycolysis rather than from mitochondrial oxidation. This metabolic shift in tumor cells was first recognized by Warburg (Warburg, 1956) and has thus become known as the Warburg effect (Gatenby and Gillies, 2004; Kim and Dang, 2006; Chen et al., 2007; Brahimi-Horn et al., 2007; Vander Heiden et al., 2009; Ganapathy et al., 2009; Mathupala et al., 2010). Given that aerobic glycolysis, which converts glucose into lactate, generates only 2 ATP whereas mitochondrial oxidation of glycolysis-derived pyruvate generates 30 ATP, it seems puzzling that tumor cells prefer the less efficient metabolic pathway to obtain energy. Tumor cells however do not suffer from ATP deficiency; in fact they generate more energy than normal cells to support their enhanced growth and proliferation. This is achieved by activation of glycolysis by several folds. It is true that mitochondrial oxidation generates more ATP than cytoplasmic glycolysis, but at the same time mitochondrial oxidation produces reactive oxygen species, which could prove to be detrimental to cells. Apparently, tumor cells recognize this negative aspect of mitochondrial oxidation and therefore opt for glycolysis as the primary source of ATP. Glycolysis does not require oxygen and does not generate reactive oxygen species. Additionally, with the generation of energy predominantly through glycolysis, tumor cells can proliferate in anerobic conditions, which often exist in solid tumors. However, the suppression of mitochondrial function in tumors is reversible. Interestingly, one of the mechanisms by which tumor cells suppress mitochondrial oxidation is by inducing PDK, thereby inactivating PDC (Semenza, 2010). Therefore, oxidation of pyruvate in mitichondria can be induced in tumor cells by reversing the cancer-associated suppression of PDC activity. Dichloroacetate does exactly that, by inhibiting PDK. Despite this rational basis for dichloroacetate as a potential anticancer drug, there is considerable controversy in the literature regarding the clinical utility of this compound for cancer treatment in humans. Even though the study by Bonnet et al (2007) demonstrated the antitumor efficacy of dichloroacetate in vitro and in vivo in animals, a recent study by Stockwin et al (2010) has shown that very high concentrations of dichloroacetate are needed to induce cell death in tumor cells and that at these concentrations the compound has no tumor cell selectivity.
Dichloroacetate is ionized and cannot pass through the plasma membrane by diffusion. This raises the question as to how this compound gets into cells and gains access to PDK within the mitochondrial matrix. To the best of our knowledge, there has been only a single report on the transport of dichloroacetate into mammalian cells, which has shown that monocarboxylate transporters in hepatocytes and Ehrlich Lettre tumor cells mediate the cellular entry of this compound (Jackson and Halestrap, 1996). Since the monocarboxylate transporters are electroneutral, most cells including tumor cells which express these transporters, may not have the ability to concentrate this drug. Recently we and others have identified a new transporter for monocarboxylates, which has substrate selectivity similar to that of the monocarboxylate transporters, but is Na+-coupled and electrogenic (Coady et al., 2004; Miyauchi et al., 2004). This transporter, known as sodium-coupled monocarboxylate transporter (SMCT1) or SLC5A8 according to the Human Genome Organization nomenclature, has the ability to concentrate its substrates against a concentration gradient because of the involvement of transmembrane Na+ gradient and membrane potential as driving forces. SLC5A8 transports acetate, propionate, butyrate, lactate, pyruvate, 3- bromopyruvate, nicotinate, β-hydroxybutyrate, and pyroglutamate (Miyauchi et al., 2004, 2010; Gopal et al., 2004, 2005; Martin et al., 2006; Thangaraju et al., 2006, 2008, 2009a). We wondered whether this highly energy-coupled transporter would accept dichloroacetate as a substrate. This issue is directly relevant to the antitumor activity of this drug because tumor cells silence this transporter by epigenetic mechanisms (Ganapathy et al., 2005, 2008, 2009; Gupta et al., 2006). Therefore, we undertook the present study to address two questions: (a) Does SLC5A8 transport dichloroacetate? (b) Does the antitumor activity of the drug depend on the expression of the transporter in tumor cells? The results of the study show that SLC5A8 is obligatory for the antitumor activity of dichloroacetate.
Results
SLC5A8 transports dichloroacetate in a Na+-coupled manner
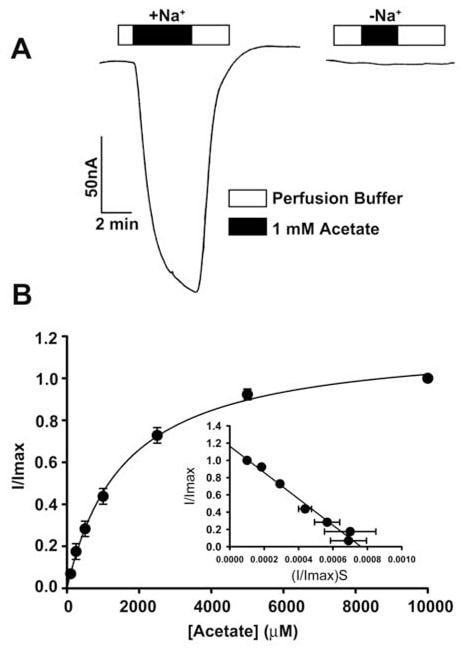
The transport of acetate and its chloro derivatives by human SLC5A8 was studied using the X. laevis oocytes expression system. The human transporter was expressed in oocytes heterologously by injection of SLC5A8 cRNA. The transport function was monitored electrophysiologically by the two-microelectrode voltage-clamp technique. SLC5A8 functions as a Na+-coupled transporter for monocarboxylates with a Na+: monocarboxylate stoichiometry of 2:1. The transport process is therefore electrogenic, associated with the transfer of one net positive charge into cells per transport cycle. The resultant depolarization of the membrane can be monitored as an inward current under voltage-clamp conditions. As can be seen in Fig. 1A, exposure of SLC5A8-expressing oocytes to acetate (1 mM) induced inward currents when monitored in the presence of Na+ in the perfusion medium (129 ± 9 nA; n = 3 oocytes). However, these currents were not observed in the absence of Na+. These data show that SLC5A8 mediates acetate transport in a Na+-coupled manner. Saturation kinetics revealed that the Michaelis constant (Kt) for the transport process was 1.6 ± 0.1 mM. After establishing the functional activity of the cloned human SLC5A8 using acetate as a positive control, we examined the transport of dichloroacetate. Exposure of SLC5A8- expressiing oocytes to dichloroacetate (0.25 mM) induced marked inward currents in the presence of Na+ (153 ± 28 nA; n = 5 oocytes) (Fig. 2A). Such currents were not observed in the absence of Na+. The transport process was saturable with a Kt of 36 ± 7 μM (Fig. 2B). Thus, the affinity of dichloroacetate for the transporter is ~45-fold higher than that of acetate. The dichloroacetate (0.25 mM)-induced currents increased as the concentration of Na+ in the perfusion medium increased (Fig. 2C). The relationship was sigmoidal, indicating involvement of more than one Na+ in the activation process. Analysis of the data according to Hill equation yielded a value of 2.1 ± 0.2 for Hill coefficient. These data show that the Na+: dichloroaceate stoichiometry for the transport process is 2:1.
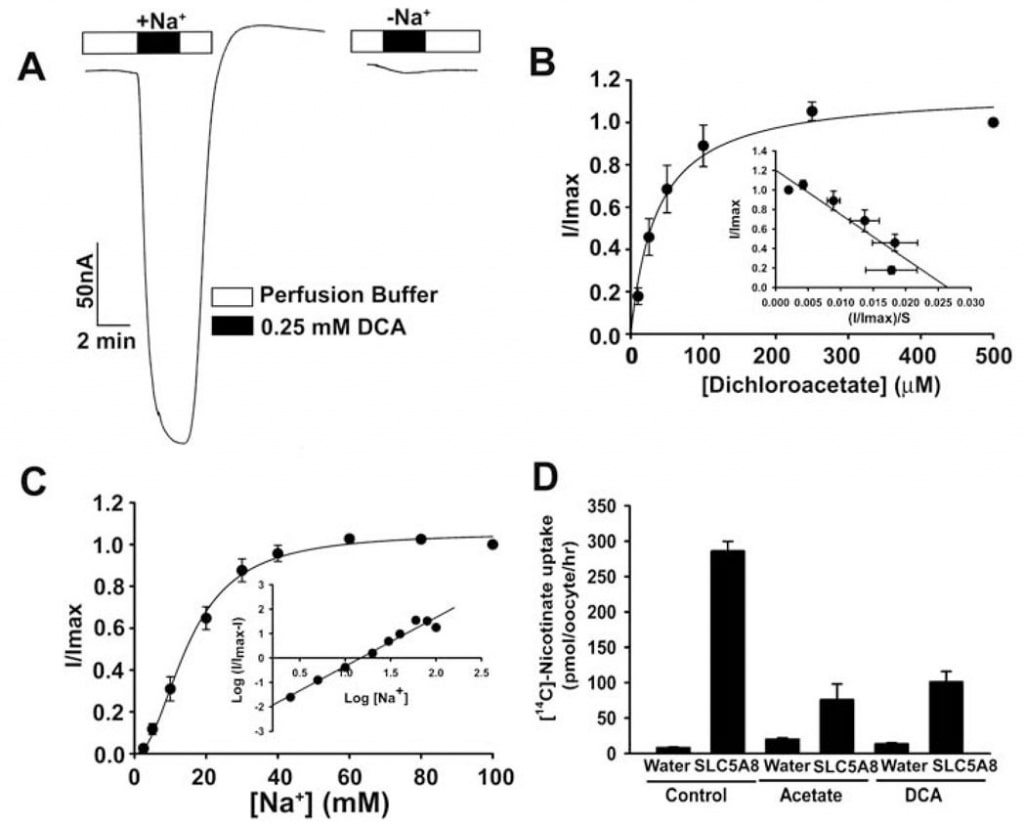
We also used an alternative method to assess the transport of dichloroacetate via SLC5A8. This method used [14C]-nicotinate as a substrate for SLC5A8. Water-injected oocytes did not express the transporter and hence were used as controls. The uptake of [14C]-nicotinate (50 μM) was 60-fold higher in SLC5A8-expressing oocytes than in water-injected oocytes (Fig. 2D). The SLC5A8-specific uptake of nicotinate was inhibited >80% in the presence of acetate (0.25 mM) or dichloroacetate (0.25 mM), indicating that acetate and dichloroacetate compete with nicotinate for uptake via SLC5A8.
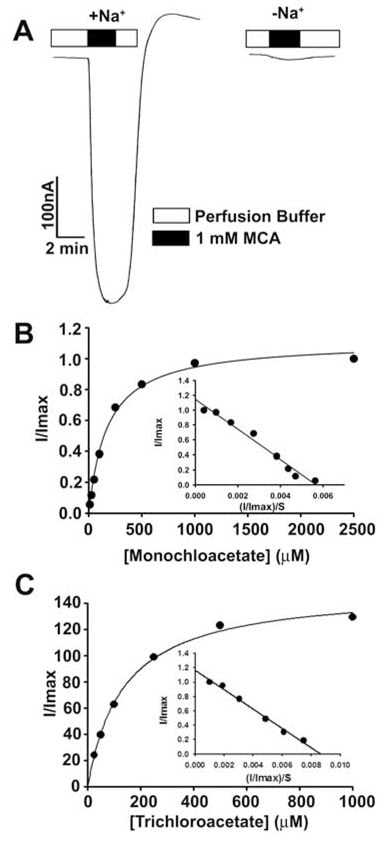
These findings showed that dichloroacetate is a high-affinity substrate for human SLC5A8. We then studied the transport of monochloroaceate and trichloroacetate for comparison (Fig. 3). Both compounds were transported via SLC5A8 in a Na+-coupled and saturable manner. The Kt was 177 ± 16μM for monochloroacetate and 134 ± 11 μM for trichloroacetate.
Dichloroacetate-induced apoptosis in cancer cells requires SLC5A8
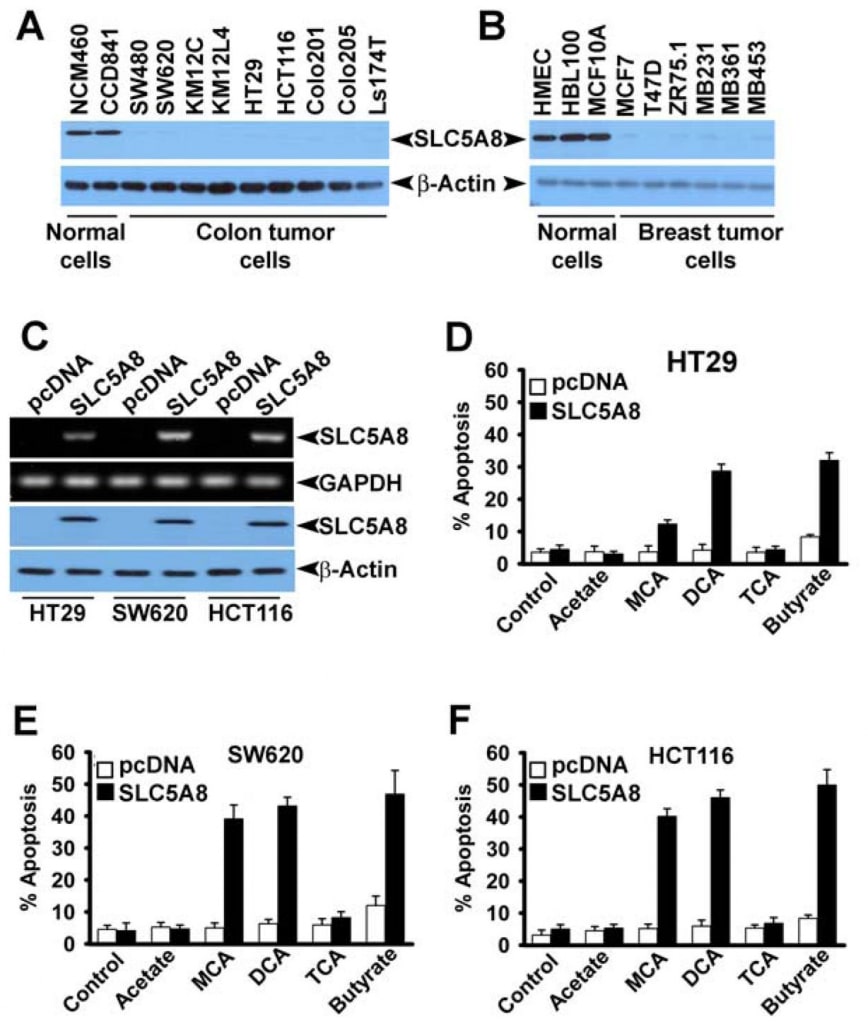
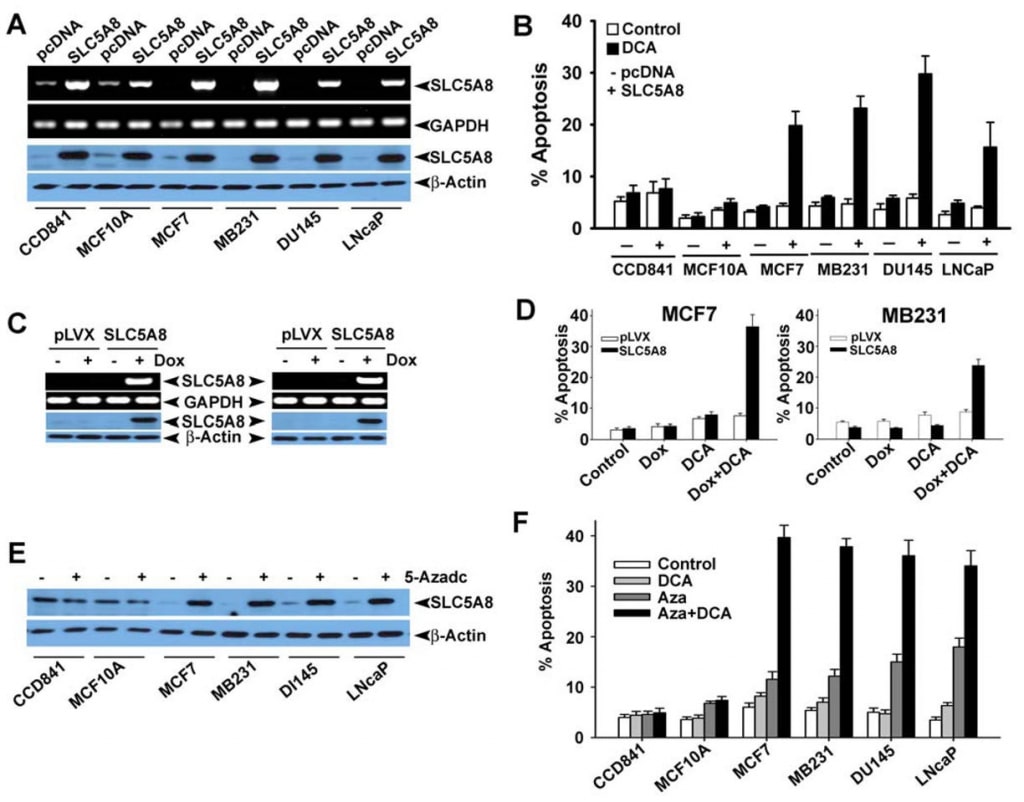
Even though several studies have shown that dichloroacetate induces apoptosis in a variety of cancer cell lines (Bonnet et al., 2007; Wong et al., 2008; Cao et al., 2008), a recent investigation was not able to confirm these findings (Stockwin et al., 2010). The studies by Bonnet et al (2007) showed that dichloroacetate at a concentration of 0.5 mM was able to induce metabolic changes specifically in cancer cells and not in normal cells. These changes included depolarization of the mitochondrial membrane, suppression of glycolysis, enhancement of mitochondrial oxidation, production of reactive oxygen species, induction of the plasma membrane potassium channel Kv1.5, and release of pro-apoptotic factors from mitochondria. Wong et al (2008) showed subsequently that dichloroacetate caused apoptosis in endometrial cancer cells, and Cao et al (2008) showed that the compound sensitized prostate cancer cells to radiation. In contrast, the studies by Stockwin et al (2010) demonstrated that even though dichloroacetate was able to induce mitochondrial depolarization and reactive oxygen species generation, these changes occurred in cancer cells as well as in normal cells. Furthermore, a very high concentration of the compound (≥25 mM) was required to induce apoptosis. Based on the findings of our present study that SLC5A8 mediates energy-coupled active entry of dichloroacetate into cells and the fact that cancer cells silence the transporter, we wondered whether the absence of the transporter in cancer cells was the reason for the lack of detectable apoptosis at low concentrations of the compound observed by Stockwin et al (2010). We addressed this question using three different human colon cancer cell lines (HCT116, SW620, and HT29). These three cell lines do not express SLC5A8 (Thangaraju et al., 2008).
We also confirmed the absence of SLC5A8 expression in human colon and breast cancer cell lines at protein level (Fig. 4A and B). We expressed SLC5A8 in HCT116, SW620, and HT29 cell lines by transient transfection of a mammalian expression construct and confirmed the expression by RT-PCR and Western blot analysis (Fig. 4C). Cells transfected with empty vector served as controls. We then exposed control and SLC5A8-expressing cells to dichloroacetate (1 mM) for 48 h and monitored apoptosis (Fig. 4D-F). The results were interesting. In control cells, which did not express the transporter, dichloroacetate did not have any significant effect. However, under identical conditions, SLC5A8-expressing cells underwent apoptosis to a marked extent. This phenomenon was seen in all three colon cancer cell lines. Monochloroacetate also behaved similar to dichloroacetate in HCT116 and SW620 cells though not in HT29 cells where the ability of monochloroacetate to induce apoptosis was significantly lower than that of dichloroacetate. Acetate and trichloroacetate were without any noticeable effect. We used butyrate as a positive control in these experiments based on our previous report that induction of apoptosis by butyrate in colon cancer cell lines is obligatorily dependent on the expression of SLC5A8 (Thangaraju et al., 2008).
We then wanted to determine if dichloroacetate-induced apoptosis exhibits tumor cell selectivity and also if the effect is seen in cancer cell lines of tissue origin other than colon. For this, we selected CCD841 and MCF10A as the representative of normal cell lines (CCD841, colon; MCF10A, mammary epithelium) and four human cancer cell lines: MCF7 (an estrogen receptor-positive breast cancer cell line), MB231 (an estrogen receptor-negative breast cancer cell line), DU145 (an androgen-insensitive prostate cancer cell line) and LNCaP (an androgen-sensitive prostate cancer cell line). As reported previously (Thangaraju et al., 2006, 2008), the normal cell lines CCD841 and MCF10A expressed detectable levels of SLC5A8, at both mRNA and protein levels (Fig. 5A). In contrast, none of the four cancer cell lines examined here expressed the transporter. The expression became evident in cancer cell lines upon transient transfection of a mammalian expression construct. In normal cells, the expression levels increased upon transfection. Using these cell lines, we compared the ability of dichloroacetate to induce apoptosis between normal and cancer cell lines (Fig. 5B). We found no significant difference in apoptosis in normal cell lines with and without treatment with dichloroacetate (1 mM). The increased levels of SLC5A8 expression also did not have any effect on the extent of apoptosis. In contrast, dichloroacetate was able to induce marked apoptosis in the four cancer cell lines, but only if the cells expressed the transporter. Without the expression of the transporter, the cancer cell lines did not undergo apoptosis upon treatment with dichloroacetate. These results show that dichloroacetate is able to cause apoptosis even at concentrations as low as 1 mM in a cancer cell-selective manner, but only if SLC5A8 is expressed. These observations were also confirmed with lenti virus-mediated stable expression of SLC5A8 (doxycylin-inducible) in two breast cancer cell lines, MCF7 and MB231 (Fig. 5C, D). It has been well established that the silencing of SLC5A8 is associated with DNA methylation and that treatment of cancer cells with DNAdemethylating agents re-actives SLC5A8 expression. Therefore, we treated normal and cancer cell lines with 5′-aza-2-deoxycytidine (5-Azadc), a DNA-demethylating agent, and the re-activation of SLC5A8 was confirmed by immunoblotting analysis (Fig. 5E). 5-Azadc treatment did not alter SLC5A8 protein expression in normal colon and mammary epithelial cells while it re-activated SLC5A8 expression in all cancer cell lines. Furthermore, treatment of these cells with 5-Azadc itself induced significant apoptosis; however, treatment of these cells with dichloroacetate (1 mM) dramatically enhanced the 5-Azadc-induced apoptosis (Fig. 5F).
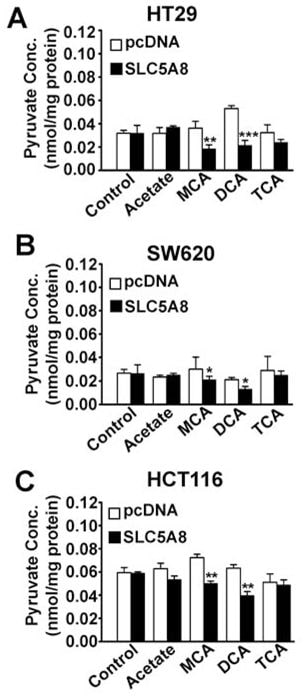
Effect of dichloroacetate on intracellular levels of pyruvate
What is the mechanism responsible for the induction of apoptosis by SLC5A8/ dichloroacetate in cancer cells? SLC5A8 is an active transporter for dichloroacetate with a Kt of 36 ± 7μM. Therefore, the compound will be concentrated in cancer cells if the transporter is expressed. This would explain the obligatory requirement for SLC5A8 for low concentrations of dichloroacetate to induce apoptosis in these cells. If the transporter is not expressed, cells may not accumulate the compound to levels sufficient enough to cause apoptosis. Once accumulated inside the cancer cells, how does dichloroacetate act to induce apoptosis? The only known mechanism of action of dichloroacetate is its ability to activate PDC through inhibition of PDK (Stacpoole et al., 1998, 2003, 2008). This would result in stimulation of mitochondrial oxidation, generation of reactive oxygen species, depolarization of mitochondrial membrane potential, and induction of apoptosis. Activation of PDC by dichloroacetate in intact cells would decrease intracellular levels of pyruvate. Based on this, we hypothesized that dichloroacetate would decrease pyruvate levels in cancer cells and that the effect would be dependent obligatorily on SLC5A8 expression. We tested this hypothesis in three different cancer cell lines (HT29, SW620, and HCT116) with and without exogenous expression of SLC5A8 (Fig. 6). The expression of the transporter by itself did not have any effect on intracellular levels of pyruvate. Dichloroacetate was able to decrease the levels of pyruvate to a significant extent in all three cancer cell lines, but only when the transporter was expressed. Interestingly, monochloroacetate also had a similar effect, again only in the presence of SLC5A8; in contrast, trichloroacetate and acetate were not able to affect pyruvate levels.
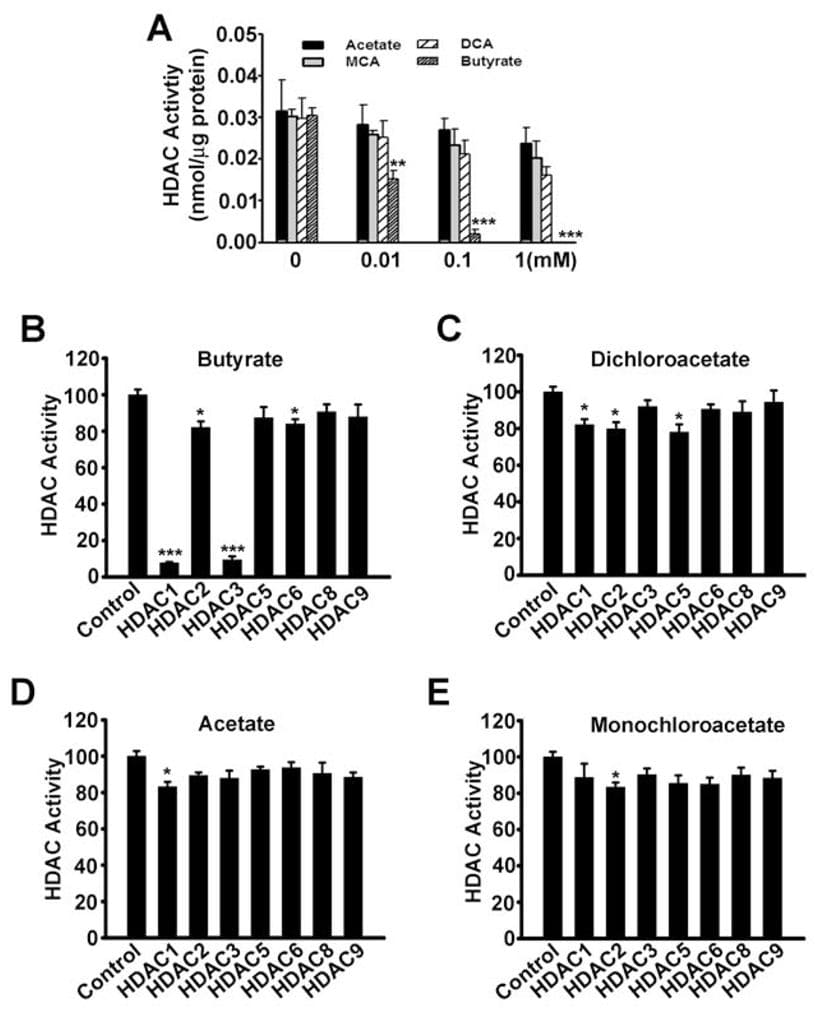
Until now the only mechanism by which SLC5A8 functions as a tumor suppressor was through its ability to concentrate inside cells butyrate and pyruvate, which are inhibitors of histone deacetylases. We have shown in the present study that dichloroacetate also induces apoptosis in cancer cells and that the effect is dependent on the expression of SLC5A8. These findings are similar to those with butyrate and pyruvate. Therefore, we wondered whether there is any involvement of inhibition of histone deacetylases in dichloroacetateinduced apoptosis in cancer cells. This seemed unlikely however because acetate is not an inhibitor of histone deacetylases (Thangaraju et al., 2006). However, we could not rule out the possibility that dichloroacetate may function as an inhibitor of histone deacetylases. Therefore, we examined the influence of acetate, monochloroacetate, dichloroacetate, and butyrate on the activity of histone deacetylases. First, we used lysates of SW620 cells as the source of histone deacetylases (Fig. 7A). Butyrate was used as a positive control in these experiments. As expected, butyrate inhibited the activity of histone deacetylases. Under identical conditions, dichloroacetate was at least 100-fold less potent compared to butyrate in inhibiting histone deacetylases. We then used recombinant human histone deacetylase isoforms to determine if specific isoforms of the enzyme could be inhibited by dichloroacetate. Our previous studies have shown that butyrate is a specific inhibitor of the histone deacetylase isoforms HDAC1 and HDAC3 (Thangaraju et al., 2009b). In the present study, we found the same with butyrate (Fig. 7B). Dichloroacetate also inhibited the isoforms HDAC1, HDAC2, and HDAC5 to a significant extent, but the potency was very low (~20% inhibition at a concentration of 1 mM) (Fig. 7C). Acetate (Fig. 7D) and monochloroacetate (Fig. 7E) did not have any effect on any of the isoforms examined in the study.
Discussion
Since the publication of the paper in Cancer Cell by Bonnet et al (2007) that showed dichloroacetate promotes apoptosis in cancer cell lines in vitro and inhibits cancer growth in mouse xenografts in vivo, there has been a steady increase in public interest in the potential utility of this compound as an anticancer drug. Dichloroaetate has been in use as a drug for treatment of congenical lactic acidosis and therefore details of the drug’s pharmacokinetics and toxicity profile have been worked out (Stacpoole at al., 1998). The recent findings that the drug may also be useful in cancer treatment have prompted many cancer patients to use this unapproved drug without their physicians’ recommendation (Pearson, 2007). Interestingly, there have been no controlled clinical trials to document the therapeutic efficacy of dichloroacetate as an anticancer drug. Since the structure of dichloroacetate cannot be patented, apparently pharmaceutical companies were not interested in developing the drug for cancer therapy through clinical trials, thus prompting the principal investigator of the original publication in Cancer Cell to raise funds for his own small clinical trials (Pearson, 2007). The results of the clinical trial have been published recently (Machelakis et al., 2010), which has shown that the drug is effective in patients with gliobastoma. Unfortunately, even though the drug was effective in vivo in the treatment of glioblastoma, high concentrations of the drug had to be used for therapeutic efficacy, which resulted in peripheral neuropathy. This was the only undesirable side effect that was limiting the use of higher dosage of the drug for greater therapeutic efficacy. There was no evidence of hematologic, hepatic, renal or cardiac toxicity. Several studies have documented neurological complications with high doses of dichloroacetate in animals and in humans (Calcutt et al., 2009; Wiemer and Sachdev, 2009; Brandsma et al., 2010). The neurological complications occur only at very high doses of dichloroacetate: 12.5 mg/kg/day for 3 months (Bonnet et al., 2007) or 15 mg/kg/day for 4 weeks (Brandsma et al., 2010). The need for high doses of dichloroacetate to be therapeutically effective in vivo agrees with most of the recently published in vitro data that show concentrations 10 mM or higher are necessary to cause apoptosis in cancer cell lines (Stockwin et al., 2010; Madhok et al., 2010; Xiao et al., 2010; Heshe et al., 2010; Sun et al., 2010).
There is convincing evidence that dichloroacetate has the ability to reverse the metabolic phenotype of cancer cells and promote their death, but the antitumor effect is seen only at very high concentrations both in vitro and in vivo. Since neurological toxicity occurs at these therapeutically effective concentrations, the utility of the compound as an anticancer drug is limited in humans. Interestingly, the only molecular target for the antitumor activity of dichloroacetate is PDK, which is inhibited by the compound at micromolar concentrations (inhibition constant, 50–100 μM) (Whitehouse et al., 1974; Cooper et al., 1974). Why is it then that high millimolar concentrations of dichloroacetate are needed in vitro to kill cancer cells? This question has to be answered in order to exploit the potential of this compound as an anticancer drug. Our present studies provide a satisfactory answer to this important question. Cancer cells do not express any transport system that can effectively transfer the compound from the extracellular medium to its intracellular target. Monocarboxylate transporters that are expressed in cancer cells transport dichloroacetate with a Kt of 0.6 mM (Jackson and Halestrap, 1996). This is a low-affinity transport process. Furthermore, monocarboxylate transporters are not highly concentrative. In addition, tumor cells generate large quantities of lactate, which is also a substrate for monocarboxylate transporters with an affinity comparable to that dichloroacetate, thus causing competition for the transport process. The present studies show that SLC5A8 transports dichloroacetate with an affinity that is considerably greater than that of monocarboxylate transporters (Kt : 36 μM versus 600 μM). Even more important is the fact that SLC5A8 is much more effective than the monocarboxylate transporters in transporting dichloroacetate because of the energizaiton of the former by a transmembrane electrochemical Na+ gradient. It is interesting to note that the affinity for the transporter changes drastically by the chlorination of the second carbon in acetate (Kt : 1572 ± 101 μM for acetate; 177 ± 16 μM for monochloroacetate; 36 ± 7 μM for dichloroacetate; 134 ± 11 μM for trichloroacetate). Our findings lead us to conclude that tumor cells are resistant to dichloroacetate-induced apoptosis because these cells do not possess effective transport systems for the drug. This is the most likely reason why very high concentrations of the compound are needed to induce cell death in tumor cells. It is also possible that the cell death observed in tumor cells at these high concentrations is not due to activation of PDC because similar effects are also seen in normal cells (Stockwin et al., 2010).
In the present study, we have shown that, if SLC5A8 is expressed in tumor cells, dichloroacetate induces cell death at a relatively lower concentration (1 mM) in a tumor cellselective manner. Under identical conditions, normal cells (CCD841 and MCF10A) are not affected. The resistance of normal cells to dichloroacetate is not due to the absence of a transport system for the drug. Normal cells express SLC5A8 constitutively, and we have shown that the cells do not undergo apoptosis with or without additional expression of the transporter. This confirms the tumor cell-specific effect of dichloroacetate. Our studies have also shown that the efficacy of dichloroacetate as an inducer of apoptosis is seen not only with colon cancer cell lines but also with breast cancer cell lines and prostate cancer cell lines. The mechanism of dichloroacetate-induced cell death in cancer cells is principally due to activation of mitochondrial oxidation of pyruvate. This is supported by the findings that intracellular levels of pyruvate in cancer cells decrease in response to treatment with dichloroacetate in a SLC5A8-dependent manner, suggesting that concentrative entry of the drug into cancer cells mediated by the exogenously expressed SLC5A8 is responsible for this effect. Dichloroacetate does not inhibit histone deacetylases, which precludes inhibition of histone deacetylases as a potential mechanism of apoptosis in tumor cells induced by the compound.
The findings that dichloroacetate induces cell death selectively in tumor cells at low concentrations but only if SLC5A8 is expressed have clinical and therapeutic significance. The ability of dichloroacetate to activate PDC via inhibition of PDK in cancer cells provides a mechanistically rational basis for the antitumor activity of the compound. But cancer cells are resistant to the drug because of the absence of an effective transporter for the drug, necessitating requirement of high concentrations of the compound to induce cell death, which unfortunately causes detrimental side effects such as neuropathy. We have demonstrated in the present study that SLC5A8 serves as an active transporter for dichloroacetate. However, since the expression of the transporter is silenced in tumor cells, how can the present findings be relevant to the potential therapeutic use of the drug? The silencing of SLC5A8 in cancer cells occurs via epigenetic mechanisms involving DNA methylation; treatment of cancer cells with 5′-azacytidine, an inhibitor of DNA methylation, re-activates the expression of the gene (Li et al., 2003; Ueno et al., 2004; Hong et al., 2005; Porra et al., 2005; Thangaraju et al., 2006; Park et al., 2007, 2008). DNA methylation plays a critical role in silencing tumor suppressor genes in a variety of cancers, and DNA methylation inhibitors hold promise as anticancer drugs (Baylin, 2005). Two compounds with DNA methylation inhibition activity are in clinical use for treatment of hematologic malignancies. These are 5′-aza-2′-deoxycytidine, also known as decitabine (trade name, Dacogen) and 5′-azacytidine (trade name, Vidaza). In vitro studies have shown that treatment of a variety of cancer cell lines with these compounds re-activates the expression of SLC5A8. We speculate that the same phenomenon would also occur in vivo. Therefore, a combination of a DNA methylation inhibitor and dichloroacetate is likely to be effective for treatment of cancer because the DNA methylation inhibitor would induce the expression of SLC5A8 in tumors, which would then effectively transport dichloroacetate into tumor cells to elicit its antitumor activity. This mode of treatment would reduce considerably the concentration of dichloroacetate necessary for in vivo efficacy as an anticancer agent, thus potentially providing tumor selectivity and also avoiding the detrimental side effects such as neuropathy. The findings of the present study provide a rational basis for such a combination therapy.
Materials and Methods
Materials
[ 14C]-Nicotinate was purchased from American Radiolabeled Chemicals (St. Louis, MO). Acetate and its chloro derivatives were obtained from Sigma (St. Louis, MO). SLC5A8 was originally cloned from human intestine (Miyauchi et al., 2004).
X. laevis oocyte expression system
Capped cRNA from human SLC5A8 cDNA (cloned in pGH19, a X. laevis oocyte expression vector) was synthesized using the mMESSAGE-mMACHINE kit (Ambion, Austin, TX). Mature oocytes from X. laevis were isolated by treatment with collagenase A (1.6 mg/ml), manually defolliculated and maintained at 18 °C in modified Barth’s medium, supplemented with 25 μg/ml gentamicin. Oocytes were injected with 50 ng of cRNA. Waterinjected oocytes served as controls. Electrophysiological studies were performed by the twomicroelectrode voltage-clamp method. Oocytes were perifused with a NaCl-containing buffer (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, pH 7.5), followed by the same buffer containing acetate or its chloro derivatives. The membrane potential was clamped at −50 mV. The differences between the steady-state currents measured in the presence and absence of substrates were considered as the substrate-induced currents. In the analysis of the saturation kinetics of substrate-induced currents, the kinetic parameter Kt (i.e., substrate concentration necessary for induction of half-maximal current) was calculated by fitting the Michaelis-Menten equation to the values of the substrateinduced currents. The Na+-activation kinetics of substrate-induced currents was analyzed by measuring the substrate-specific currents in the presence of increasing concentrations of Na+. The data for the Na+-dependent currents were analyzed according to the Hill equation to determine the Hill coefficient (the number of Na+ ions involved in the activation process). Since expression levels varied significantly from oocyte to oocyte, kinetic analyses were done by normalizing the expression levels. This was done by taking the maximally induced SLC5A8-specific current in each kinetic experiment in individual oocytes as 1. Each experiment was repeated with 3 or 4 different oocytes. The activity of the heterologously expressed SLC5A8 in oocytes was also monitored by [14C]-nicotinate uptake. The concentration of nicotinate in these experiments was 50 μM and the time of incubation was 1 h.
The protocol for the use of frogs in these experiments was approved by the Institutional Animal Care and Use Committee.
Ectopic expression of SLC5A8 in cultured cell lines
Cells were seeded in 35-mm culture dishes and cultured in the absence of pyruvate. Cells were transfected with pcDNA or human SLC5A8 cDNA using Fugene 6 and Opti-MEM. pEGFP-N1 was used for co-transfection to determine transfection efficiency. After 24 h, cells were treated with or without acetate or its chloro derivatives (1 mM) for 24 h. For FACS analysis, cells were fixed in 50% ethanol, treated with 0.1% sodium citrate, 1 mg/ml RNase A, and 50 μg/ml propidium iodide, and then subjected to fluorescence-activated cell sorting (FACS; FACS Caliber, Becton Dickinson) analysis.
Generation of SLC5A8-expressing stable cell lines
We generated SLC5A8-expressing stable clones in two cell lines, MCF7and MDA-MB231, to conditionally express SLC5A8 by placing SLC5A8 cDNA under the control of Tet dependant promoter (Tet-On Advanced, Clontech). The system comes with two elements, the regulator vector (pLVX-Tet-On Advanced) and the response vector (pLVX-Tight-Puro). Briefly, recombinant lenti virus was produced by co-transfection into 293FT cells the regulator vector (pLVX-Tet-On Advanced) and three other helper vectors, pLP-1, pLP-2 and pVSVG (Invitrogen) using Lipofectamine 2000 transfection reagent. Lenti viral supernatant was harvested 72 h post-transfection and filtered through 0.45 μm membrane. MCF7 and MDA-MB231 cells were infected for 24 h with lenti virus in medium containing 8 μg/ml polybrene and cultured for an additional 48 h. Cells were selected with G418 (2 mg/ml) and the expression of rTet R protein was checked with Western blot. SLC5A8 was subcloned into pLVX-Tight-Puro and SLC5A8 pLVX-Tight-Puro was transduced into MCF7 and MDA-MB231 cells expressing the rTetR protein with the same procedure described above. The cells were selected with 2 μg/ml puromycin and maintained in medium containing Tetfree FBS, 0.25 μg/ml puromycin, and 250 μg/ml G418. The induction of SLC5A8 mRNA on addition of doxycycline was confirmed by RT-PCR.
Re-activation of SLC5A8 expression by DNA-demethylating agent
Cells were seeded in 10-cm dishes at very low density (0.5 × 106 cells/dish) and cultured in respective medium without sodium pyruvate. After 24 h, cells were treated with 5′-Azadc (2 μg/ml) for 72 h. The medium was replaced with fresh medium containing 5′-Azadc for every 24 h. Following the treatment with 5′-Azadc, cells were treated with DCA (1 mM) for 48 h. After the treatment, cells were harvested and processed for protein extraction as well as for FACS analysis.
Reverse transcription-PCR
RNA prepared from normal and cancercell lines wasused for semi-quantitative RT PCR. RNA (2 μg) was reverse-transcribed into cDNA using the GeneAmp PCR system (Roche). The PCR primers for specific genes were designed based on the nucleotide sequences available in GenBank. Reverse transcription-PCR (RT-PCR) was repeated twice with each RNA sample. GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) was used as the internal control.
Western blot analysis
Fifty micrograms of protein was fractionated by SDS-PAGE, and the fractionated proteins were transferred onto a nitrocellulose membrane (Whatman GmbH). Membranes were blocked with 5% non-fat dry milk and then exposed to anti-SLC5A8 primary antibody (Cat. # ARP44110, Aviva System Biology) at 4 °C overnight, followed by treatment with appropriate secondary antibodies. Proteins were visualized by ECL SuperSignal Western System (GE Healthcare).
Analysis of apoptosis
Cells were fixed in 50% ethanol, treated with 0.1% sodium citrate, 1 mg/mL RNase A, and 50 μg/mL propidium iodide, and subjected to fluorescence-activated cell sorting (FACS, Becton Dickinson) analysis.
Measurement of HDAC activity
A commercially available kit (Biovision, Mountain View, CA) was used to determine HDAC activity. When the cell lysate of the colon cancer cell line SW620 was used as the source of HDAC activity, 100 μg of protein in the lysate was added to the enzyme assay in the presence or absence of acetate or its chloro derivatives (1 mM). The activity of recombinant human HDAC isoforms was also measured using the same kit. The recombinant HDAC isoforms were purchased from Cayman Chemical Company.
Measurement of intracellular levels of pyruvate
Cells were seeded in 35-mm culture dishes and cultured in the absence of pyruvate. Cells were transfected with pcDNA or human SLC5A8 cDNA using Fugene 6 and Opti-MEM. After 24 h, cells were treated with or without acetate or its chloro derivatives (1 mM) for 24 h. Cell lysates were then used for measurement of pyruvate using a commercially available kit (Biovision, Mountain View, CA).
Acknowledgments
The work described here is supported in part by the National Institutes of Health grant CA131402.
REFERENCES
1 Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005; 2:S4–S11. [PubMed: 16341240]
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondriaK + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37–51. [PubMed: 17222789]
2Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia signaling controls metabolic demand. Curr Opin Cell Biol. 2007; 19:223–229. [PubMed: 17303407]
2Brandsma D, Dorlo TP, Haanen JH, Beijnen JH, Boogerd W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J Neurol. 2010 In press.
2Calcutt NA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, et al. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009; 68:985–993. [PubMed: 19680144]
3Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008; 68:1223–1231. [PubMed: 18465755]
4Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007; 39:267–274. [PubMed: 17551814]
5Coady MJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. The tumour suppressor gene SLC5A8 expresses a Na+-monocarboxylate cotransporter. J Physiol. 2004; 557:719–731. [PubMed: 15090606]
6Cooper RH, Randle PJ, Denton RM. Regulation of heart muscle pyruvate dehydrogenase kinase. Biochem J. 1974; 143:625–641. [PubMed: 4462746]
7Ganapathy V, Gopal E, Miyauchi S, Prasad PD. Biological functions of SLC5A8, a candidate tumor suppressor. Biochem Soc Trans. 2005; 33:237–240. [PubMed: 15667316]
8Ganapathy V, Thangaraju M, Gopal E, Itagaki S, Miyauchi S, Prasad PD. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008; 10:193–199. [PubMed: 18446519]
9Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009; 121:29–40. [PubMed: 18992769]
10Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4:891– 899. [PubMed: 15516961]
11Gopal E, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin PM, et al. Expression of Slc5a8 in kidney and its role in Na+-coupled transport of lactate. J Biol Chem. 2004; 279:44522–44532. [PubMed: 15322102]
12Gopal E, Fei YJ, Miyauchi S, Zhuang L, Prasad PD, Ganapathy V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by Slc5a8, a member of the Na+/glucose cotransporter gene family. Biochem J. 2005; 388:309–316. [PubMed: 15651982]
13Gupta N, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006; 78:2419– 2425. [PubMed: 16375929]
14Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol. 2010 In press.
15Hong C, Maunakea A, Jun P, Bollen AW, Hodgson JG, Goldenberg DD, et al. Shared epigenetic mechanisms in human and mouse gliomas inactivate expression of the growth suppressor SLC5A8. Cancer Res. 2005; 65:3617–3623. [PubMed: 15867356]
16Jackson VN, Halestrap AP. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2′, 7′-bis (carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996; 271:861–868. [PubMed: 8557697]
17Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006; 66:8927–8930. [PubMed: 16982728]
19Li H, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci USA. 2003; 100:8412–8417. [PubMed: 12829793]
20Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cellcycle arrest in colorectal cancer cells. Br J Cancer. 2010; 102:1746–1752. [PubMed: 20485289]
21Martin PM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, et al. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J Neurochem. 2006; 98:279–288. [PubMed: 16805814]
22Mathupala SP, KOYH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010; 1797:1225–1230. [PubMed: 20381449]
23Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008; 99:989–994. [PubMed: 18766181]
24Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010; 2:31–34.
25Miyauchi S, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor downregulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem. 2004; 279:13293–13296. [PubMed: 14966140]
26Miyauchi S, Gopal E, Babu E, Sonne SR, Kubo Y, Umapathy NS, et al. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim Biophys Acta. 2010; 1798:1164–1171. [PubMed: 20211600]
27Park JY, Zheng W, Kim D, Cheng JQ, Kumar N, Ahmad N, et al. Candidate tumor suppressor gene SLC5A8 is frequently down-regulated by promoter hypermethylation in prostate cancer. Cancer Detect Prev. 2007; 31:359–365. [PubMed: 18037591]
28Park JY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, et al. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas. 2008; 36:e32–e39. [PubMed: 18437076]
29Pearson H. Cancer patients opt for unapproved drug. Nature. 2007; 446:474–475. [PubMed: 17392750]
30Porra V, Ferraro-Peyret C, Durand C, Selmi-Ruby S, Giroud H, Berger-Dutrieux N, et al. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations is classical papillary thyroid carcinomas. J Clin Endocrinol Metab. 2005; 90:3028–3035. [PubMed: 15687339]
31Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010; 20:51–56. [PubMed: 19942427]
32Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab Rev. 1998; 30:499–539. [PubMed: 9710704]
33Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003; 43:683–691. [PubMed: 12856382]
34Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008; 60:1478–1487. [PubMed: 18647626]
35Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate (DCA) selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010 in press.
36Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 2010; 120:253–260. [PubMed: 19543830]
37Thangaraju M, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res. 2006; 66:11560–11564. [PubMed: 17178845]
38Thangaraju M, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short-chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. J Gastrointest Surg. 2008; 12:1773–1782. [PubMed: 18661192]
39Thangaraju M, Karunakaran S, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Transport via SLC5A8 with subsequent inhibition of histone deacetylases HDAC1 and HDAC3 underlies the antitumor activity of 3-bromopyruvate. Cancer. 2009a; 115:4655–4666. [PubMed: 19637353]
40Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009b; 417:379–389. [PubMed: 18789002]
41Ueno M, Toyota M, Akino K, Suzuki H, Kusano M, Satoh A, et al. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004; 25:134– 140. [PubMed: 15361710]
42Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–1033. [PubMed: 19460998]
43Warburg O. On the origin of cancer cells. Science. 1956; 123:309–314. [PubMed: 13298683]
44Weimer LH, Sachdev N. Update on medication-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2009; 9:69–75. [PubMed: 19080756]
45Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974; 141:761–774. [PubMed: 4478069]
46Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008; 109:394–402. [PubMed: 18423823]
47Xiao L, Li X, Niu N, Qian J, Xie G, Wang Y. Dichloroacetate (DCA) enhances tumor cell death in combination with oncolytic adenovirus armed with MDA-7/IL-24. Mol Cell Biochem. 2010; 340:31–40. [PubMed: 20165905]